Evolutionary genomics of bacteria
The increasing availability of bacterial genomes has opened the possibility for addressing fundamental questions of the evolutionary biology of bacteria:
- What does the genome wide variation can tell us about the demographic history of bacterial species?
- How has the gene composition of the genomes changed over time?
- How has the acquisition of genes (outside of the common drug resistance genes) via lateral gene transfer contributed to the adaptation of bacterial species to new niches?
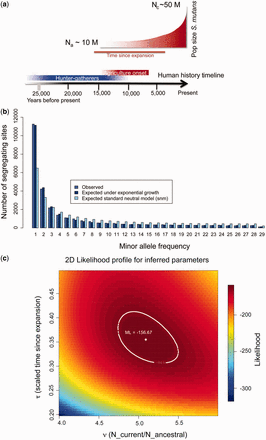
How did Streptococcus mutans evolved to become the main cavities causing bacteria
We have been working on applying some of the recent advances in population genomic techniques to understand the evolutionary history of microbes. An example of the kind of studies we have done revealed that Streptococcus mutans populations, the cavities causing bacteria, have been expanding, most likely after the origin of agriculture (Cornejo et at 2013). More importantly, we have found that there is a subset of genes, unique to this species, which are functionally involved in the metabolism of sugars and resistance to low pH environments, selective pressures that the bacteria had to face after the change of diet that accompanied the development of agriculture.
In my lab we are interested in applying the same analysis framework to other moderately recombining bacterial species (commensal and pathogenic) to try understand how populations of bacteria have changed in time and how the changes in human population have modified the population demographics of its pathogens and vice versa.
Ecology and evolution of bacteriocins
The evolution of an autolysin in Streptococcus pneumoniae and the ecology responsible for the evolution and maintenance of a self-killing agent
 Agents that kill the organisms that produce them or other, genetically identical members of their populations are intriguing puzzles for ecologists and evolutionary biologists. How can such “self-killing” agents, which, on a first consideration appear to be a considerable disadvantage to the organisms that produce them evolve and be maintained by natural selection? One possibility we have suggested is that these agents can be maintained if during the interaction of different bacterial populations or species, the producing species kill their competitors more than they kill themselves (Cornejo et al. 2009).
Agents that kill the organisms that produce them or other, genetically identical members of their populations are intriguing puzzles for ecologists and evolutionary biologists. How can such “self-killing” agents, which, on a first consideration appear to be a considerable disadvantage to the organisms that produce them evolve and be maintained by natural selection? One possibility we have suggested is that these agents can be maintained if during the interaction of different bacterial populations or species, the producing species kill their competitors more than they kill themselves (Cornejo et al. 2009).
This work, at the intersection of experiments and mathematical modeling has also opened the door for an interesting hypothesis about the maintenance of self-killing agents with a set of testable hypotheses: if the toxin producing bacterium kills others more than it kills itself, then such a system could potentially be maintained. Nevertheless, as with the advantage of recombination, this mechanism engenders a cost to the organism and its action is frequency or density-dependent, making the question of its origin and maintenance very controversial.
Biology of recombination in bacteria
 In bacteria, recombination is a rare event and not part of its reproductive process. Nevertheless, the importance that Horizontal Gene Transfer (HGT in a broad sense) plays in the adaptive evolution of bacteria species is increasingly recognized. Because of its ability to acquire genes and genetic elements from other organisms, the pace of adaptive evolution in bacteria (i.e. invasion of new niches, competition against other bacteria or acquisition of resistance to antibiotics and other environmental stresses) need not to be limited by the amount of standing variation in the population.
In bacteria, recombination is a rare event and not part of its reproductive process. Nevertheless, the importance that Horizontal Gene Transfer (HGT in a broad sense) plays in the adaptive evolution of bacteria species is increasingly recognized. Because of its ability to acquire genes and genetic elements from other organisms, the pace of adaptive evolution in bacteria (i.e. invasion of new niches, competition against other bacteria or acquisition of resistance to antibiotics and other environmental stresses) need not to be limited by the amount of standing variation in the population.
But then, we can ask ourselves: why do bacteria maintain an intricate machinery for homologous gene recombination? What is the impact of homologous gene recombination on the maintenance and accessibility of standing genetic variation in populations where sex is not frequent and not linked to reproduction? These are questions that are not well understood. Our work suggests that, once established in the population, the ability to recombine can be maintained in the population at the expense of its costs.
Nevertheless, because of the frequency and density-dependent nature of horizontal gene transfer in bacteria, the benefits of recombination are not that clear when this ability is rare. We have performed computational studies to generate testable hypothesis about the evolution of recombination in bacterial populations that we plan on pursuing in the future using Streptococcus pneumoniae as a model system.
 Another important aspect of recombination in bacteria is understanding the factors that limit or promote the acquisition of foreign DNA into the cell. My previous work, done in collaboration with Daniel Rozen (now at the University of Leiden, the Netherlands) has shown that, contrary to some hypothesis proposed in the microbial molecular genetics literature, the polymorphism in the competence system responsible for inducing the uptake of DNA does not maintain genetically differentiated populations. This research has also open the door to interesting findings on the evolution of the polymorphism of two component systems (receptor – signal peptides) and we plan on pursuing additional experimental work to understand how polymorphism can be maintained by purifying selection via compensatory mutations. The interesting bit is that while sequence-wise the competence system presents strong similarities with self-compatibility systems in plants, the mechanisms maintaining polymorphism in the population are radically different.
Another important aspect of recombination in bacteria is understanding the factors that limit or promote the acquisition of foreign DNA into the cell. My previous work, done in collaboration with Daniel Rozen (now at the University of Leiden, the Netherlands) has shown that, contrary to some hypothesis proposed in the microbial molecular genetics literature, the polymorphism in the competence system responsible for inducing the uptake of DNA does not maintain genetically differentiated populations. This research has also open the door to interesting findings on the evolution of the polymorphism of two component systems (receptor – signal peptides) and we plan on pursuing additional experimental work to understand how polymorphism can be maintained by purifying selection via compensatory mutations. The interesting bit is that while sequence-wise the competence system presents strong similarities with self-compatibility systems in plants, the mechanisms maintaining polymorphism in the population are radically different.
Additional work performed by Benjamin Evans, a former student of Danny Rozen has shown results that are consistent with our original assessment of the impact of the polymorphism on the population substructure of S. pneumoniae.
Metagenomic analysis and host associations

There is an increased recognition that microbial communities associated with humans can modulate the outcomes of health and disease. We are interested in studying the association between microbial communities and host genetics to better predict health and disease outcomes.
One question that is driving the research in our lab is the accurate and repeatable identification and representation of microbial communities. This is a problem when beta-diversity of microbial communities is such that communities have a significant overlap and uncertainty in the identification is problematic. This problem is relatively prevalent in the identification of microbial community types from the human vagina. Five main community types (CSTs) have been identified and four of them are dominated by Lactobacilli species (figure to the left). We have developed a method that we call Microbionn that improves significantly on existing methodologies. We will be releasing a pre-print and access to the github soon. In the figure below you can see the confusion matrix of one existing method (Valencia) compared to the one obtained with Microbionn. You can see a lower incidence of off-diagonal values (mis-identifications) in our method.

In my lab we are interested in applying the same analysis framework to other moderately recombining bacterial species (commensal and pathogenic) to try understand how populations of bacteria have changed in time and how the changes in human population have modified the population demographics of its pathogens and vice versa.
Relevant publications
- Xia, C., Wang, M., Yin , C., Cornejo, O.E., Hulbert, S.H., Chen. X. (2018) Genomic insights into host adaptationbetween the wheat stripe rust pathogen(Puccinia striiformisf. sp.tritici) andthe barley stripe rust pathogen(Puccinia striiformisf. sp.hordei) BMC Genomics 19(1):664.
- Xia C, Wang M, Yin C, Cornejo O.E., Hulbert S, Chen X. (2018) Genome sequence resources for the wheat stripe rust pathogen (Puccinia striiformis f. sp. tritici) and the barley stripe rust pathogen (Puccinia striiformis f. sp. hordei). Mol Plant Microbe Interact. doi: 10.1094/MPMI-04-18-0107-A.
- Xia, C., Wang, M., Cornejo, O.E., Jiwan, D.A., See, D.R., Chen, X. (2017). Secretome characterization and correlation analysis reveal putative pathogenicity mechanisms in the wheat stripe rust fungus Puccinia striiformis f. sp. tritici. Frontiers in Microbiology 2017;00-12. doi: 10.3389/fmicb.2017.02394.
- Miller EL, Evans BA, Cornejo OE, Roberts IS, Rozen DE. (2017). Pherotype polymorphism in Streptococcus pneumoniae has no obvious effects on population structure and recombination. Genome Biology and Evolution 9(10): 2546–2559. doi.org/10.1093/gbe/evx188
- Cornejo OE*, Hickey RJ, Suzuki H, Forney LJ*. (2017). Focusing the diversity of Gardnerella vaginalis through the lens of ecotypes. Evolutionary Applications 2017;00:1–13. https://doi.org/10.1111/eva.12555 (*co-corresponding authors)
-
Pepperell CS, Casto AM, Kitchen A, Granka JM, Cornejo OE, Holmes EC, Birren B, Galagan J, Feldman MW (2013) The role of selection in shaping diversity of natural M. tuberculosis populations. PLoS Pathogens. 9(8): e1003543. doi: 10.1371/journal.ppat.1003543
-
Cornejo OE, Lefébure T, Bitar PD, Lang P, Richards VP, Eilertson K, Do T, Beighton D, Zeng L, Ahn SJ, Burne RA, Siepel A, Bustamante CD, Stanhope MJ. (2013) Evolutionary and population genomics of the cavity causing bacteria Streptococcus mutans. Molecular Biology and Evolution. 30(4): 881-893. [Highlighted in Science: “How Sweet it is: genes show how bacteria colonized human teeth” by Ann Gibbons]
-
Cornejo OE, McGee L, Rozen DE. (2010) Polymorphism in the competence peptide does not limit recombination in Streptococcus pneumoniae. Molecular Biology and Evolution. 27: 694-702.
-
Levin BR, Cornejo OE. (2009) The Population and Evolutionary Dynamics of Homologous Gene Recombination in Bacteria. PLoS Genetics 5(8): e1000601. doi:10.1371/journal.pgen.1000601.
-
Cornejo OE, Rozen DE, May RM, Levin BR. (2009) Oscillations in Continuous Culture Populations of Streptococcus pneumoniae: Population Dynamics and the Evolution of Clonal Suicide. Proceedings of the Royal Society B. 276(1659):999-1008.